By Adam Kinsella (School of Mathematics & Physics)
Supervisors: Dr Matt Booth, Mr Chris Dickens
Semiconductors are an incredibly valuable, highly-sought-after technology with a wide variety of uses, the most pressing of which is solar panels; as the climate crisis becomes increasingly urgent, research into the electrical properties of different semiconductor materials is more relevant than ever.
Copper Aluminium Sulfide (CuAlS2) is one such material; our research involved doping pure CuAlS2 with various amounts of Iron (Fe) to form CuAl16-xFexS2, where x represents the number of Iron atoms. The conductive properties of these doped systems was then explored through DFT (Density Functional Theory) simulation.
As the project began, I read up on some of the different simulation techniques available within DFT, and taught myself the mathematical theory behind the method we used (PBE). I was unfamiliar with Fortran, the coding language used; I spent some time going through online tutorials and literature to learn more about the language. This also involved adapting knowledge of other coding languages from previous computing experience to help with Fortran programming.
Two doping methods were used:

-
-
- Replacing Aluminium with Iron (“Direct”)
- Replacing an Aluminium atom with Copper, then the nearest Copper atom with Iron (“Indirect”)
-
We tested how these different techniques impacted the conductive properties of the material. For each method, VMD software was used to visualise the system (see Figure 1); from here, we used the distances between atoms to determine which atoms to replace. I enjoyed using VMD, as it provided a lot of different ways of viewing the material.
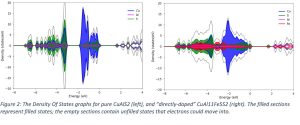
Once the simulations had been run, I co-wrote Python scripts with Chris Dickens, one of my supervisors, to calculate and graph the “Density Of States” (DOS) of each system (see Figure 2). These graphs give information on the number of “free” states – i.e., states available for electrons to move into. Whilst I was familiar with basic semiconductor physics from the Condensed Matter Physics module of my undergraduate degree, the density of states concept was complicated, and it took a while (and a lot of background reading!) to wrap my head around it.
On top of the DOS graphs, I wrote a Python script to calculate the formation energies of each system (the difference in energy before vs after the Iron defects were added). Calculating these energies allowed for the 2 methods of doping to be compared; the lower the formation energy, the easier it is to add new defects. The equation used to calculate the formation energy was quite complicated, so it took a few days and some discussion with my supervisors to get the program working.
Over the course of the project, I’ve definitely learned a lot, and I was surprised by how many different areas I ended up learning about – I’ve gained knowledge in computing/programming, in DFT simulation, in the physical concepts behind semiconductors and in collaborative research. I’ve enjoyed the process of working alongside my supervisors and being directly involved in the decisions made on this project and believe my experience will aide me in a wide variety of areas in the future.
*To view the research poster for this project, please click on the thumbnail below: